A DEFINITION
Molecular modelling is a combination of bioinformatical and biophysical techniques to represent and visualize the shapes of molecules in space, and to simulate the way they move and behave.

A DEFINITION
Molecular modelling is a combination of bioinformatical and biophysical techniques to represent and visualize the shapes of molecules in space, and to simulate the way they move and behave.
Knowing a protein’s 3D structure helps researchers understand not only how the protein works on the atomic scale but also how it moves and interacts with other proteins or molecules such as drugs for example.
Proteins are too small (100 to 1,000 angstroms) to be seen with classical microscopy techniques.
Building on the positions of all the atoms that make up a protein, molecular modelling is a technique used for ‘visualizing’ a protein’s 3D structure.
HOW CAN YOU 'SEE' THE ATOMS IN A PROTEIN?
Here are two techniques that are used to determine ‘experimentally’ the positions of a protein’s atoms.
The protein is crystallized and its crystal structure analyzed using x-ray diffraction.
The position of every single atom is then determined thanks to mathematical analysis.
This technique has been used since the 1960s.
Nuclear Magnetic Resonance (NMR) is a technique used to analyse proteins in solution. With NMR, researchers are able to determine the relative positions of each hydrogen and carbon atom and hence, indirectly, a protein’s 3D structure.
Cryogenic electron microscopy (cryo-EM) is a more recent technique. With cryo-EM, researchers can study the 3D structure of proteins in biological samples frozen at -185°C.
In the 1980s, Prof. Jacques Dubochet developed a method known as vitrification. Vitrification brings the temperature of water far down without changing its molecular structure. He shared the 2017 Nobel Prize in chemistry for this discovery which is the basis of cryo-EM. The 3D structure of haemoglobin (3.4 angstroms) was determined by using this technique (source).

When the position of each atom in every single amino acid and the links between the atoms are known, scientists can design the 3D structure of a protein thanks to molecular modelling.
Here are 2 representations of the 3D structure of the BRAF protein, which is composed of 766 amino acids (and 11,815 atoms).
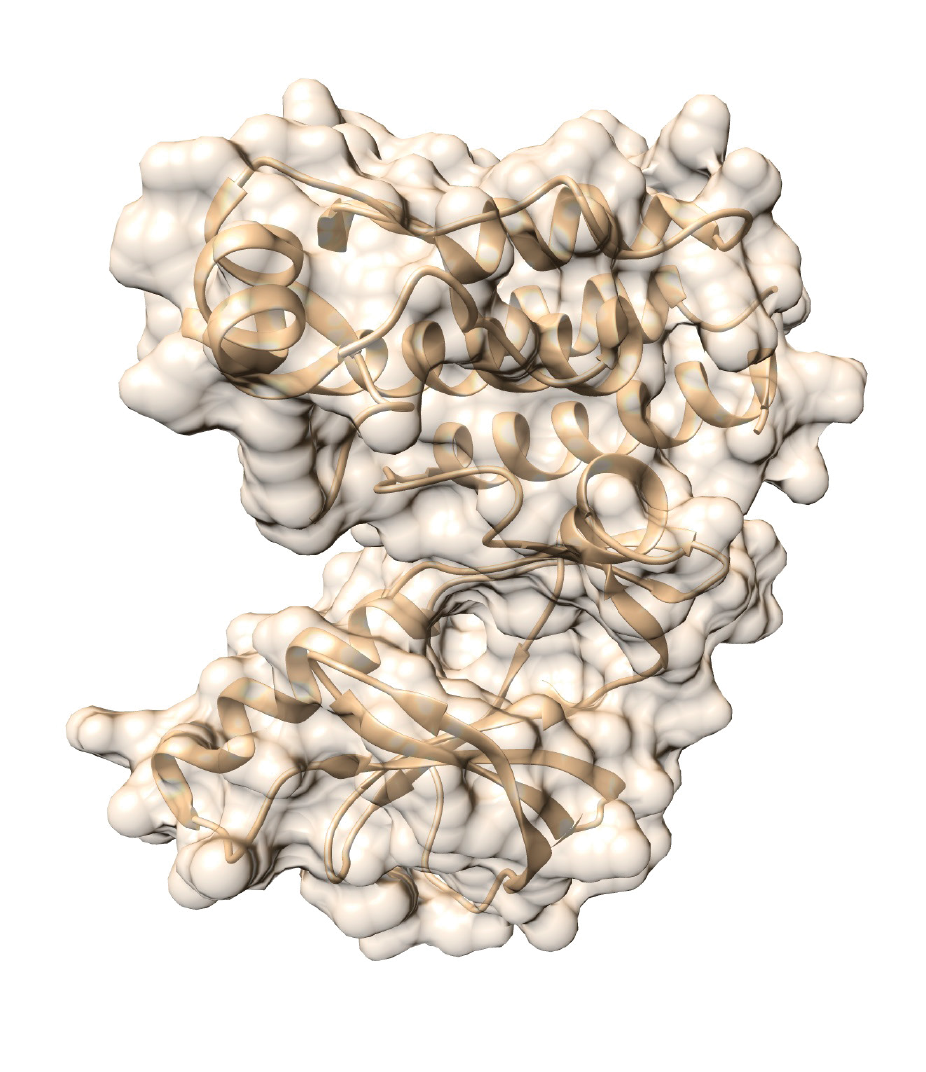
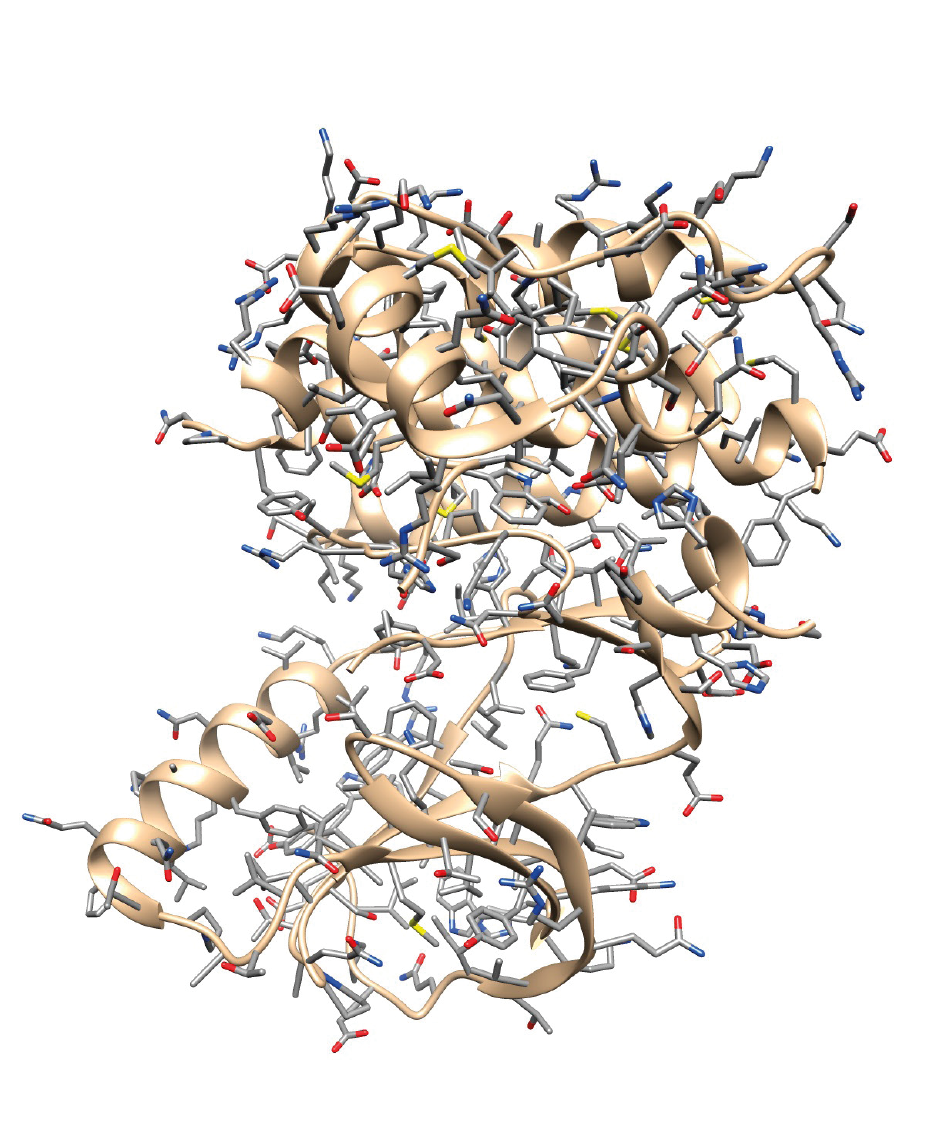

About two thirds of the structure of human proteins have not been determined « experimentally » (source UniProtKB).
Thanks to molecular modelling, it is possible to predict their 3D structure on the basis of the 3D structure of proteins that are already known and to which they are similar.

Thanks to molecular modelling, researchers can predict the effect that one or several amino-acid changes could have on a protein’s 3D structure.
Consequently, it is possible to predict the effect a mutation could have on a protein’s activity and/or on its capacity to interact with a drug.
In particular, molecular modelling is used when there is no biological or medical information available on a given mutation.
This is the case for about 25% of patients taken care of by the Molecular Tumor Board du Réseau d’Oncologie Romand.
THE BRAF V600E EXAMPLE
The BRAF protein with the V600E mutation has lost its ability to control cell division. The drug vemurafenib targets the mutated protein, blocking its activity.
Molecular modelling helps to visualize and simulate the effect the V600E mutation would have when the drug interacts with the mutated protein.
The drug inserts itself into the BRAF V600E protein where it is physically and chemically more stable. As a result, the drug’s affinity is higher for the BRAF protein with the mutation (BRAF V600E) as opposed to the normal protein.
Vemurafenib is used to treat patients whose cancer cells have the BRAF V600E mutation.
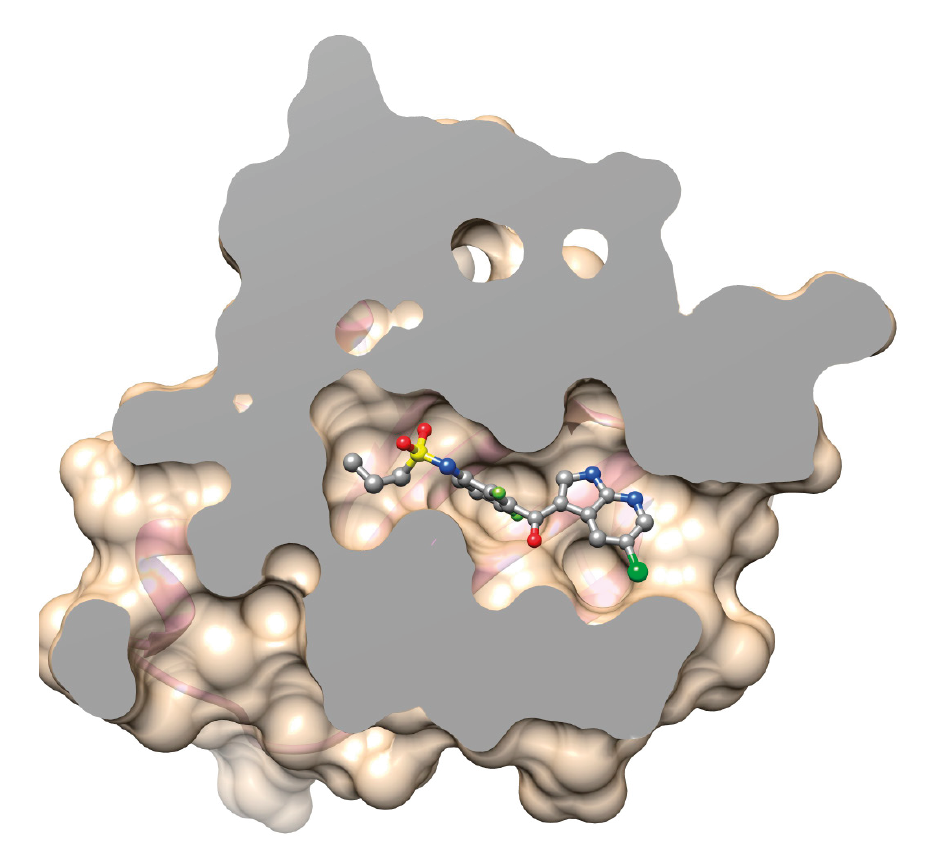
Normal BRAF
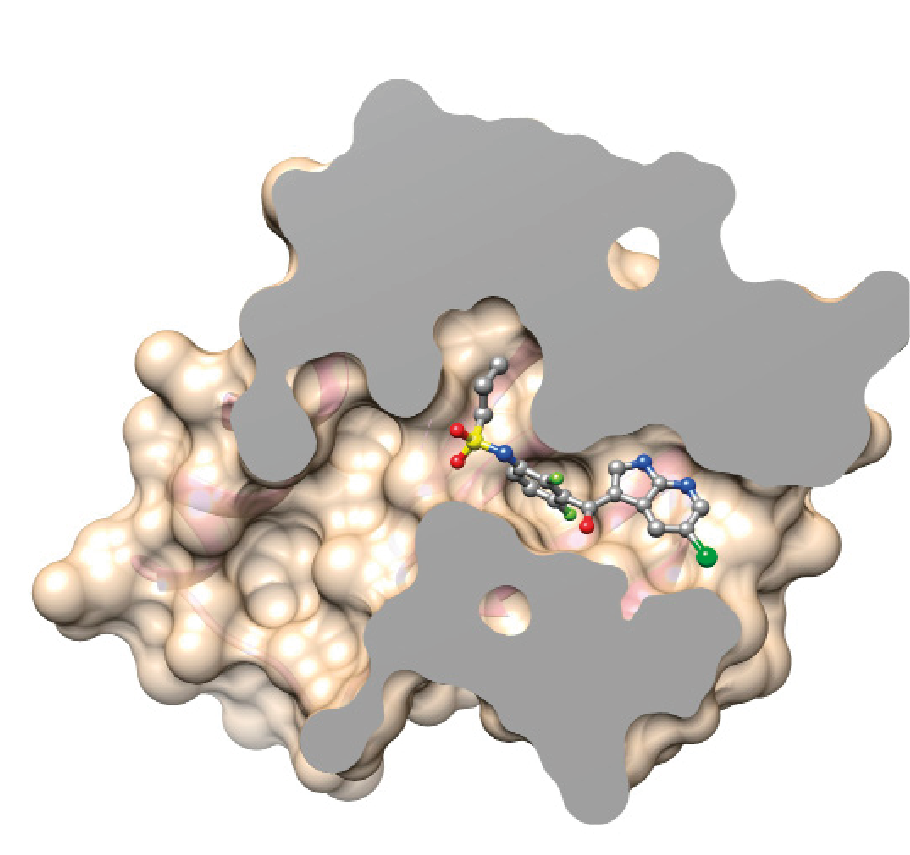
BRAF V600E

The techniques of molecular modelling are also used to design novel drugs.
Once the 3D structure of a targetable protein is known, it is possible to design a drug likely to insert itself into the protein’s functional site. More often than not, the goal is to block the activity of the protein responsible for the disease.
AN ANTI-HIV DRUG: A HISTORICAL EXAMPLE
Marketed in 1987, saquinavir was one of the first drugs that was designed using bioinformatical programs.
Saquinavir specifically targets one of HIV’s proteins, known as a protease. This particular protease is needed for HIV’s replication and survival.
Researchers determined the 3D structure of the protease. They then designed a drug likely to insert itself into the protein’s functional site and block the protein’s activity.
And it worked!

Saquinavir is one of the drugs currently used in HIV/AIDS triple therapy.
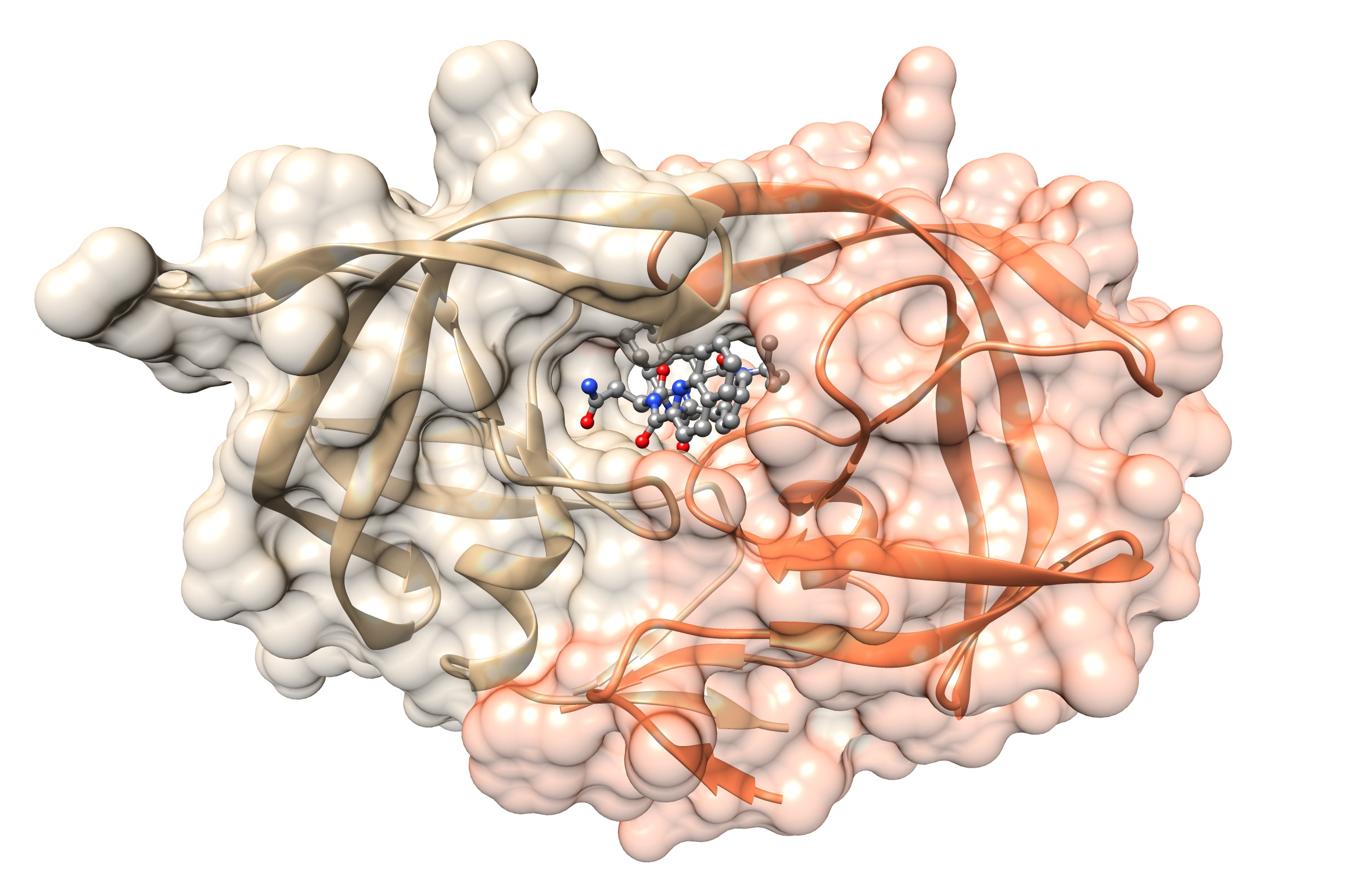
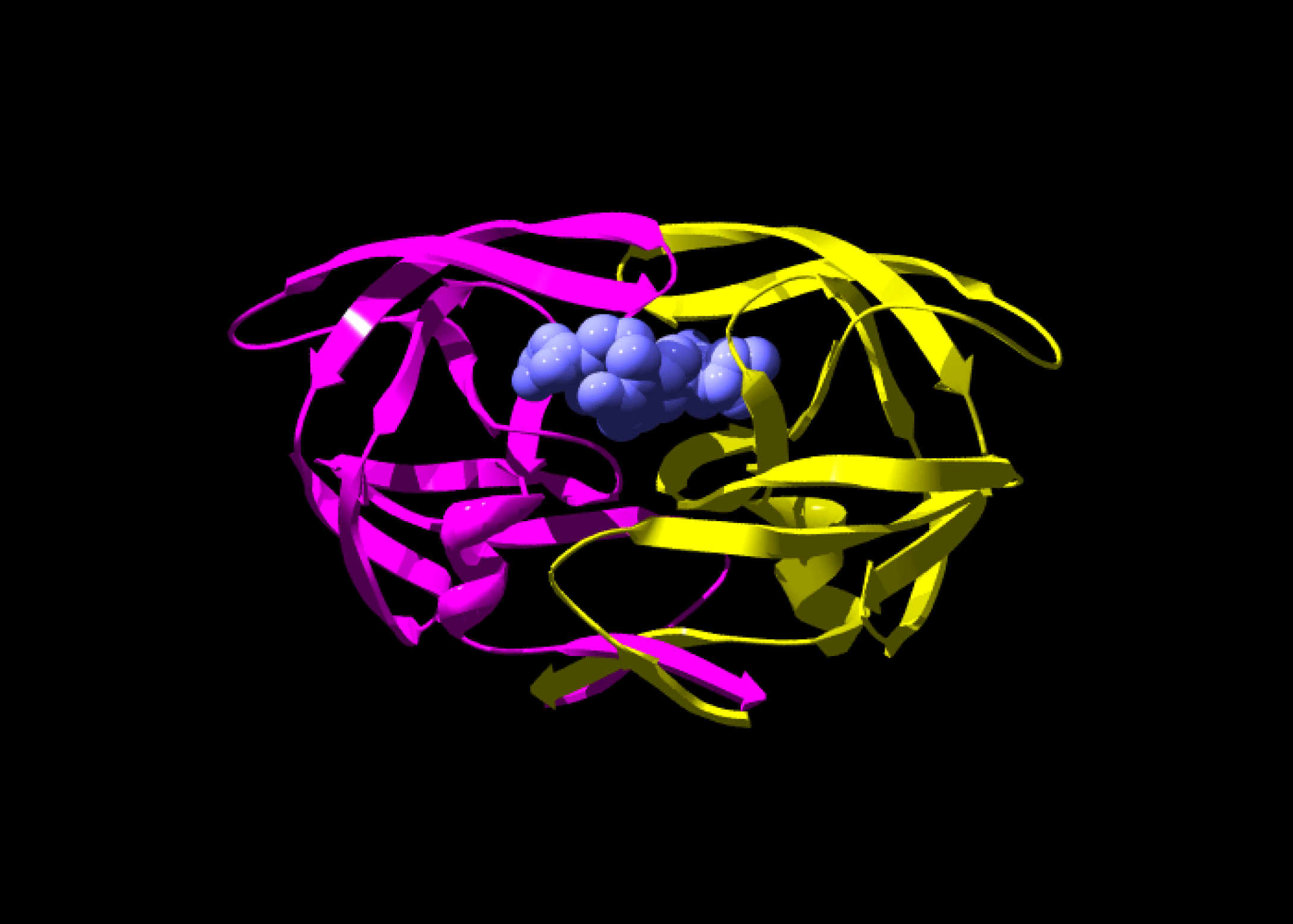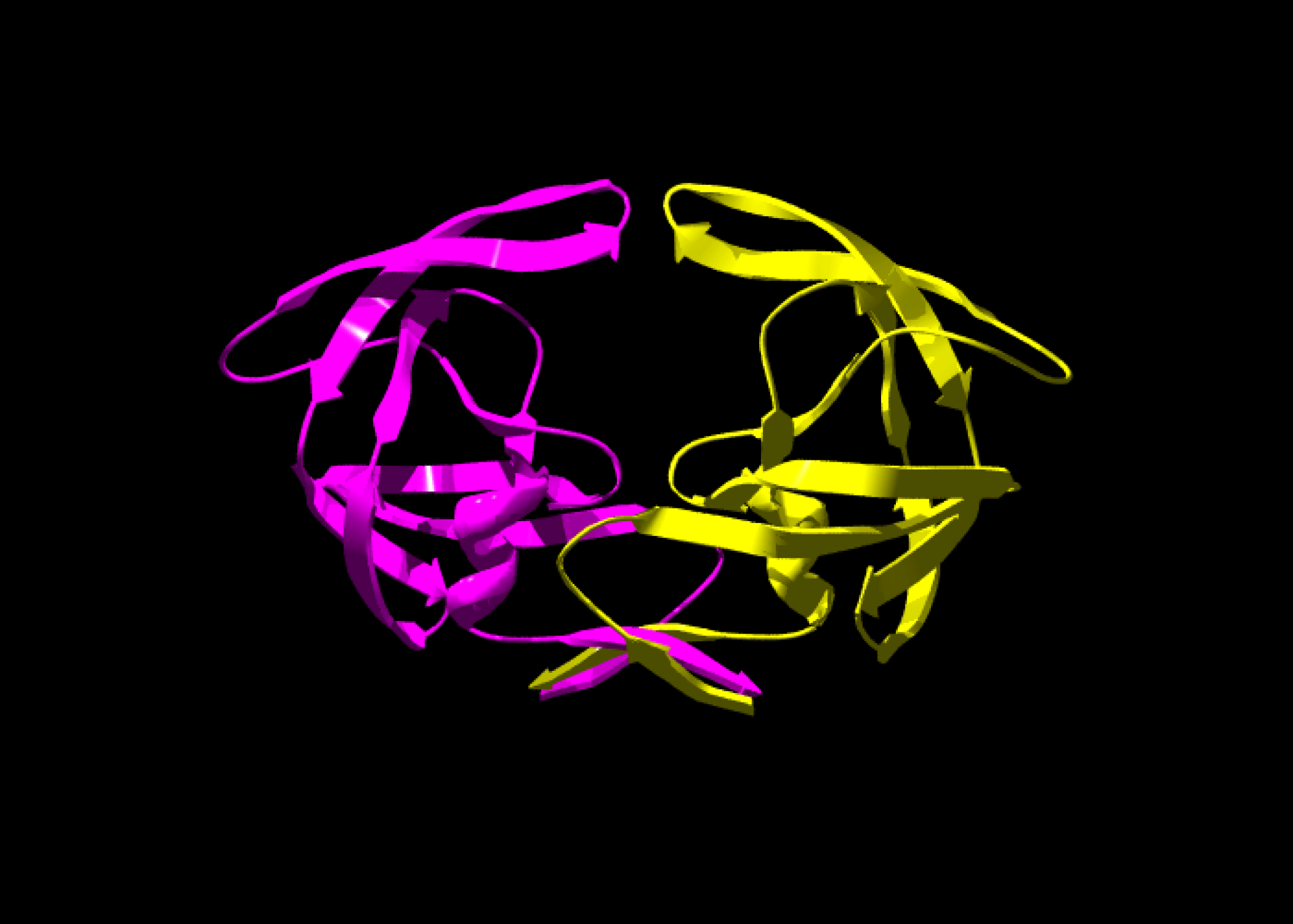
Another representation of the 3D structure of the HIV protease with or without its inhibitor: the drug saquinovir.

